Quantum molecular mechanics
A noniterative procedure for the fast Ab Initio calculation of closed shell systems
Gustavo Laureano Coêlho Moura, and Alfredo Mayall Simas
Journal of Computational Chemistry, 33(9), 958-969, 2012
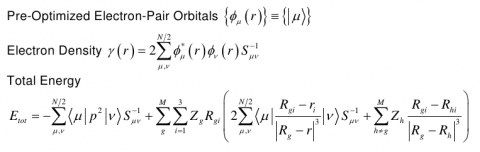
QMM is a novel molecular mechanics ppt method based, not on classical or semiclassical force fields, but entirely on quantum mechanics and localized non-orthogonal electron-pair orbitals.
In QMM, the orbitals are not fixed for each chemical bond. They vary according to variations in geometry. Consequently, QMM appears to correctly describe the response of the electron density to variations in geometry and thus takes into account polarization and charge transfer effects between atoms involved in a chemical bond.
Moreover, assuming that the QMM spatial orbitals are fully optimized for the system under consideration, we showed that the calculation of the total energy can actually be carried out without resorting to any two-electron integrals.